Meningioma: Clinical Presentation, Diagnostic Evaluation, Management, and Outcomes – A Case Report
OthersPage Navigation
Abstract
Meningiomas are the most common primary intracranial tumors, arising from the meningothelial cells of the arachnoid layer. They are typically slow-growing and benign, accounting for approximately 30–40% of all primary brain tumors. Despite their benign histology in most cases, meningiomas can cause significant neurological morbidity due to mass effect and compression of adjacent neural structures.
We report the case of a 48-year-old female presenting with progressive headaches and intermittent visual disturbances over 8 months. Neuroimaging revealed a well-defined extra-axial mass consistent with a meningioma. The patient underwent surgical resection followed by regular monitoring. Histopathology confirmed a WHO Grade I meningioma. Postoperative recovery was favorable, with resolution of symptoms and no evidence of recurrence at 6-month follow-up.
This case underscores the importance of early recognition, appropriate imaging, and timely intervention in achieving optimal outcomes in meningioma patients.
Introduction
Meningiomas are extra-axial tumors originating from arachnoid cap cells of the meninges. They are generally benign, with approximately 80–85% classified as World Health Organization (WHO) Grade I tumors. However, atypical (Grade II) and anaplastic (Grade III) variants exhibit more aggressive behavior and higher recurrence rates.
These tumors commonly occur in middle-aged to elderly individuals and demonstrate a higher prevalence in females, suggesting a hormonal influence in tumor development. Common locations include the cerebral convexities, parasagittal region, sphenoid wing, and posterior fossa.
Clinically, meningiomas may remain asymptomatic for long periods and are often incidentally detected. Symptomatic cases typically present with headaches, seizures, focal neurological deficits, or visual disturbances depending on tumor location.
Advancements in neuroimaging, surgical techniques, and adjuvant therapies have significantly improved patient outcomes. However, recurrence and long-term monitoring remain important considerations in management.
Case Report
Patient History
A 48-year-old female presented with the following complaints:
- Progressive, dull, holocranial headache for 8 months
- Occasional blurred vision
- Episodes of dizziness
- No history of seizures or loss of consciousness
The headache was insidious in onset, non-radiating, and not relieved by over-the-counter analgesics. The patient reported gradual worsening in intensity over time.
There was no history of trauma, fever, vomiting, or focal weakness. Medical history was unremarkable, and there was no known history of malignancy.
Family history was non-contributory.
Clinical Examination
General Examination
- Patient conscious, alert, and oriented
- Vitals stable
- No pallor, cyanosis, or edema
Neurological Examination
- Cranial nerves: Mild visual field defect noted
- Motor system: Normal tone and power
- Sensory system: Intact
- Reflexes: Normal
- Cerebellar signs: Absent
Clinical Evaluation
Differential Diagnosis
Based on clinical presentation, the following were considered:
- Meningioma (most likely)
- Glioma
- Metastatic brain tumor
- Pituitary adenoma (if parasellar involvement)
- Vestibular schwannoma (depending on location)
The slow progression and absence of systemic malignancy favored a benign intracranial tumor such as meningioma.
Investigations
Neuroimaging
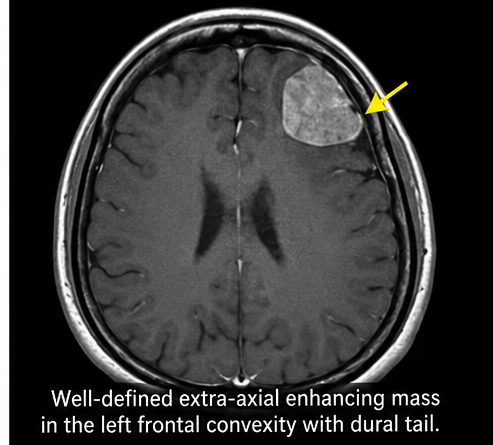
Magnetic Resonance Imaging (MRI) Brain:
- Well-circumscribed extra-axial mass
- Location: Left frontal convexity
- Size: 3.5 × 2.8 cm
- Homogeneous contrast enhancement
- Dural tail sign present
- Mild surrounding edema
Computed Tomography (CT) Scan:
- Hyperdense lesion with calcifications
- No evidence of hemorrhage
Laboratory Tests
- Complete blood count: Within normal limits
- Renal and liver function tests: Normal
- Coagulation profile: Normal
Diagnosis
A provisional diagnosis of intracranial meningioma was made based on characteristic imaging findings, including a well-circumscribed extra-axial mass with homogeneous contrast enhancement and the presence of a dural tail sign on MRI. The lesion’s location, defined margins, and associated mild peritumoral edema further supported the likelihood of a benign meningioma rather than other intracranial neoplasms. Additionally, the absence of features suggestive of high-grade malignancy such as irregular borders, necrosis, or significant invasion into adjacent brain parenchyma nreinforced this working diagnosis. These radiological characteristics, in conjunction with the patient’s clinical presentation of slowly progressive symptoms, were highly consistent with a typical meningioma.
Management and Outcome
Management Strategy
The treatment approach focused on:
- Symptom relief
- Complete tumor resection
- Prevention of recurrence
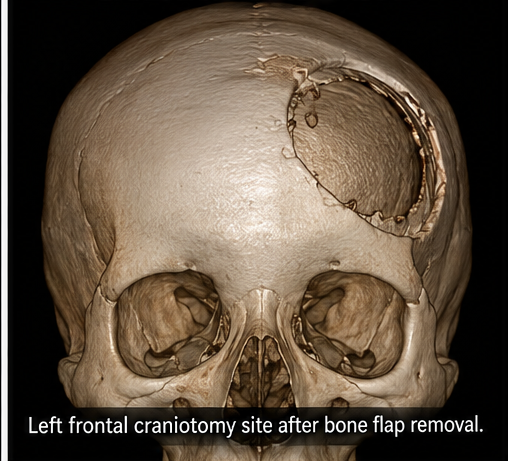
Surgical Management
The patient underwent:
- Left frontal craniotomy
- Complete excision of tumor (Simpson Grade I resection)
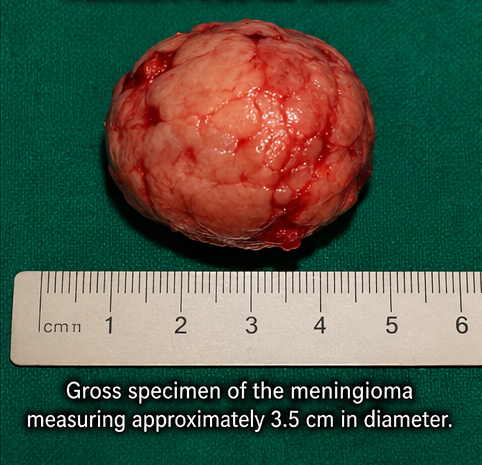
Intraoperative findings:
- Well-encapsulated tumor
- Firm consistency
- Clear plane of cleavage from brain tissue
Histopathological Examination
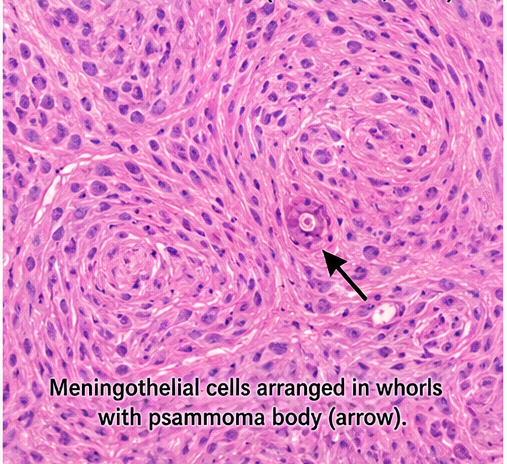
- Tumor composed of spindle-shaped meningothelial cells
- Whorl formation observed
- No atypia or mitotic activity
Diagnosis: WHO Grade I meningioma
Postoperative Course
- Uneventful recovery
- Headache resolved within 1 week
- Vision improved gradually
- No neurological deficits
Follow-Up
At 1 Month
- No residual symptoms
- Surgical site healed
At 3 Months
- MRI showed no residual or recurrent tumor
- Patient resumed normal activities
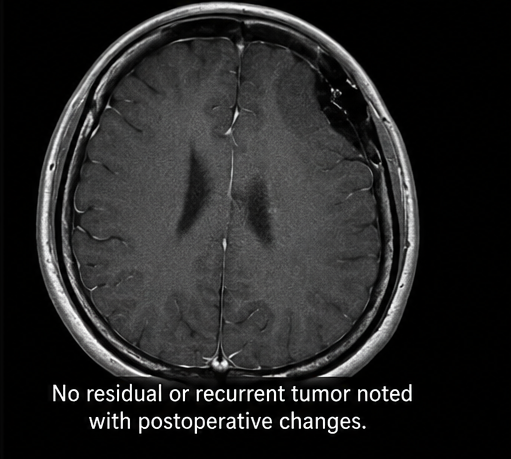
At 6 Months
- No recurrence
- Excellent functional recovery
Discussion
Pathophysiology
Meningiomas arise from arachnoid cap cells and are typically slow-growing tumors. The pathogenesis involves genetic mutations, most commonly involving the NF2 gene on chromosome 22.
Hormonal factors, particularly progesterone receptors, are thought to contribute to tumor growth, explaining the higher incidence in females.
Etiology and Risk Factors
Key risk factors include:
- Female gender
- Increasing age
- Ionizing radiation exposure
- Genetic syndromes (e.g., Neurofibromatosis type 2)
- Hormonal influences
Epidemiology
- Account for 30–40% of primary brain tumors
- More common in females (2:1 ratio)
- Peak incidence: 40–60 years
- Mostly benign
Clinical Manifestations
Symptoms depend on tumor location and size:
- Headache (most common)
- Seizures
- Visual disturbances
- Focal neurological deficits
- Cognitive changes
In this case, headache and visual disturbance were predominant.
Diagnostic Considerations
Diagnosis relies primarily on imaging:
- MRI brain with contrast (gold standard)
- CT scan (useful for calcifications)
- Histopathology for definitive diagnosis
Key imaging feature:
- Dural tail sign (characteristic of meningioma)
Treatment Considerations
Surgical Resection
- First-line treatment for symptomatic tumors
- Goal: Complete excision
- Simpson grading predicts recurrence
Radiotherapy
- Indicated in:
- Incomplete resection
- Recurrent tumors
- Atypical/anaplastic meningiomas
Medical Therapy
- Limited role
- Hormonal therapy and targeted agents under investigation
Emerging Therapies
- Targeted molecular therapies
- Immunotherapy
- Stereotactic radiosurgery (Gamma Knife)
- Anti-angiogenic agents
Complications
Potential complications include:
- Tumor recurrence
- Neurological deficits
- Seizures
- Postoperative infections
Prognosis
Prognosis depends on:
- Tumor grade
- Completeness of resection
- Location
- Patient age
WHO Grade I meningiomas have an excellent prognosis with low recurrence rates after complete resection.
Conclusion
Meningiomas are common intracranial tumors with predominantly benign behavior but significant potential for neurological impact. This case highlights the importance of early diagnosis through imaging and effective surgical management.
Complete surgical resection remains the cornerstone of treatment, with excellent outcomes in most cases. Long-term follow-up is essential due to the risk of recurrence.
Advances in molecular biology and targeted therapies hold promise for improved management of complex and recurrent cases in the future.
References
- Wiemels J, Wrensch M, Claus EB. Epidemiology and etiology of meningioma. https://pubmed.ncbi.nlm.nih.gov/24139766/
- Ostrom QT, et al. CBTRUS Statistical Report: Primary brain tumors. https://pubmed.ncbi.nlm.nih.gov/33123732/
- Louis DN, et al. WHO Classification of CNS Tumors. https://pubmed.ncbi.nlm.nih.gov/33288970/
- StatPearls. Meningioma Overview. https://www.ncbi.nlm.nih.gov/books/NBK560538/
- Goldbrunner R, et al. EANO guidelines for meningioma. https://pubmed.ncbi.nlm.nih.gov/31014271/
- Sughrue ME, et al. Meningioma recurrence factors. https://pubmed.ncbi.nlm.nih.gov/21394103/
- Whittle IR, et al. Meningiomas: Pathogenesis and management. https://pubmed.ncbi.nlm.nih.gov/15231236/
Read more such content on @ Hidoc Dr | Medical Learning App for Doctors
Recommended News For You
Recommended Articles For You
Featured News
Featured Articles
Featured Events
Featured KOL Videos
1.
Electronic Sepsis Alerts; Reducing Plaques in Coronary Arteries
2.
Ivonescimab Tops Pembrolizumab in PD-L1-Positive, Advanced NSCLC
3.
Hereditary cancer has a rare and underreported cause.
4.
New imaging guidelines for head and neck cancers, a step toward practice change
5.
BMTs that are "half-matched" are effective in treating severe sickle cell disease.
1.
Oncolytic Adenoviruses Targeting PD-L1: Advancing Cancer Immunotherapy and Tumor Control
2.
Personalized Cancer Vaccines: The Next Frontier in Precision Oncology
3.
Essential Updates in Hematology in Daily Practice
4.
The Predictive Power of Theranostics in Palliative Neuroendocrine Tumor Management
5.
Importance of Early Detection in Oncology
1.
Asian Symposium on Advancement in Hematology and Oncology
2.
Asian Symposium on Advancement in Hematology and Oncology
3.
Asian Symposium on Advancement in Hematology and Oncology
4.
International Cancer Conference
5.
Asian Symposium on Advancement in Hematology and Oncology
1.
A Comprehensive Guide to First Line Management of ALK Positive Lung Cancer - Part VII
2.
Expert Group meeting with the management of EGFR mutation positive NSCLC - Part I
3.
Current Scenario of Cancer- The Incidence of Cancer in Men
4.
Untangling The Best Treatment Approaches For ALK Positive Lung Cancer - Part IV
5.
A New Era in Managing Cancer-Associated Thrombosis

Address :
Hidoc Dr. Inc. | Delaware C Corp | 1309 Coffeen Ave. Suite 1200, Sheridan WY, 82801
Phone :
+1-415-463-3094
Email :
anishagadia@hidoc.co
© Copyright 2026 Hidoc Dr. Inc.
Terms & Conditions - LLP | Inc. | Privacy Policy - LLP | Inc. | Account Deactivation

To get started please enter your email ID
Welcome to Hidoc Dr.
Join to enhance your clinical skills and gain specialized in-depth medical knowledge