Choroidal Coloboma: Clinical Presentation, Diagnostic Evaluation, and Long-Term Visual Outcomes - A Case Report
OthersPage Navigation
Abstract
Choroidal coloboma is a rare congenital ocular anomaly resulting from incomplete closure of the embryonic fissure during early ocular development. It primarily affects the posterior segment of the eye and may involve the choroid, retina, retinal pigment epithelium, and optic nerve. The condition is often diagnosed incidentally but can be associated with significant visual impairment and sight-threatening complications such as retinal detachment and choroidal neovascularization.
We report a case of unilateral choroidal coloboma in a young adult presenting with long-standing visual impairment. This case highlights the clinical presentation, diagnostic challenges, multimodal imaging findings, differential diagnoses, management strategies, and long-term follow-up considerations. The report emphasizes the importance of early recognition, patient counseling, and regular ophthalmic surveillance to prevent vision-threatening complications.
Introduction
Coloboma is a congenital ocular defect caused by failure of fusion of the embryonic (choroidal) fissure between the fifth and seventh weeks of gestation. Depending on the extent and site of involvement, coloboma may affect the iris, ciliary body, choroid, retina, and optic nerve. Choroidal coloboma refers to posterior segment involvement and represents one of the more visually significant forms of this developmental anomaly.
The condition typically involves the inferonasal quadrant of the fundus and may be unilateral or bilateral. Choroidal coloboma can occur as an isolated finding or as part of multisystem congenital syndromes such as CHARGE syndrome, cat eye syndrome, and trisomy 13. Visual prognosis varies widely and depends on factors such as macular involvement, optic nerve coloboma, and secondary complications.
Although there is no definitive curative treatment for choroidal coloboma, early diagnosis and long-term monitoring play a crucial role in preserving visual function. This case report presents a typical clinical scenario of unilateral choroidal coloboma and discusses diagnostic evaluation, management principles, and prognostic considerations.
Case Report
Patient History
A 24-year-old man presented to the ophthalmology outpatient clinic with complaints of decreased vision in the right eye since early childhood. The visual impairment was nonprogressive and not associated with pain, redness, photophobia, flashes, or floaters. The patient denied any history of ocular trauma, surgery, or acute visual loss.
There was no history of systemic illness, developmental delay, congenital anomalies, or hearing impairment. The patient was born at term following an uncomplicated pregnancy and delivery. There was no reported history of maternal infection, medication exposure, or substance use during pregnancy. Family history was negative for ocular or congenital disorders.
Clinical Examination
On examination, best-corrected visual acuity (BCVA) was 6/36 in the right eye and 6/6 in the left eye. Color vision testing revealed mild impairment in the right eye, while contrast sensitivity was reduced compared to the contralateral eye.
Anterior segment examination using slit-lamp biomicroscopy showed normal eyelids, conjunctiva, cornea, anterior chamber, iris, and lens in both eyes. No iris coloboma or other anterior segment anomalies were noted. Pupillary reactions were normal, with no relative afferent pupillary defect.
Intraocular pressure measured by applanation tonometry was within normal limits in both eyes.
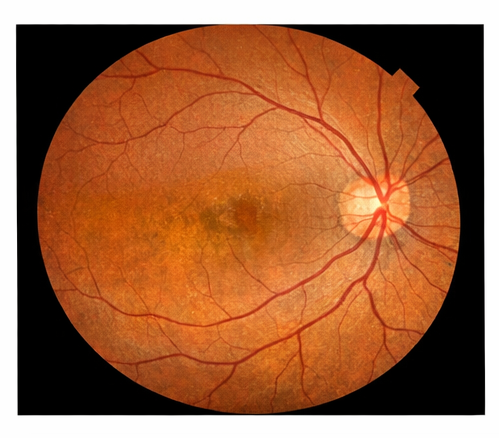
Dilated fundus examination of the right eye revealed a large, well-defined, pale excavated lesion involving the inferonasal quadrant of the posterior pole, extending from the optic disc margin toward the peripheral retina.
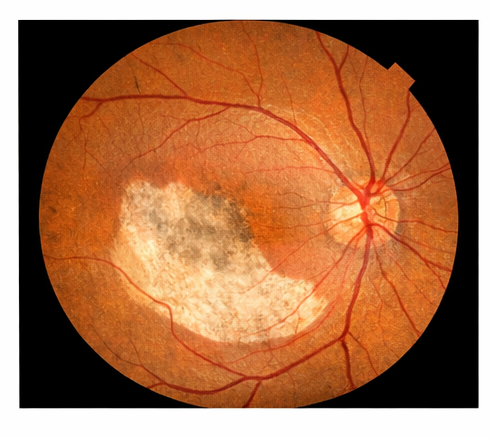
The lesion demonstrated absence of normal choroidal vasculature and retinal pigment epithelium, consistent with choroidal coloboma.
The macula was partially involved, explaining the reduced visual acuity. The optic disc appeared mildly dysplastic but without definitive optic nerve coloboma.
Fundus examination of the left eye was unremarkable.
Clinical Evaluation
Differential Diagnosis
Based on clinical findings, the following differential diagnoses were considered:
- Choroidal coloboma
- Congenital chorioretinal atrophy
- Healed chorioretinal scar due to intrauterine infection
- Toxoplasmosis scar
- Posterior staphyloma
The congenital onset, typical inferonasal location, well-circumscribed margins, and characteristic appearance favored a diagnosis of choroidal coloboma.
Investigations
Imaging Studies
Color fundus photography was performed to document the size and extent of the lesion.
Optical coherence tomography (OCT) of the right eye revealed complete absence of normal retinal layers and choroid within the colobomatous area, with abrupt thinning of adjacent retinal tissue. The foveal architecture was disrupted, correlating with reduced visual acuity.
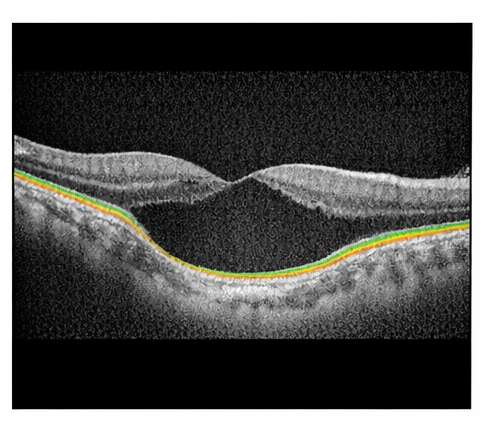
Enhanced depth imaging OCT demonstrated marked choroidal thinning at the margins of the coloboma. There was no evidence of subretinal fluid or choroidal neovascular membrane.
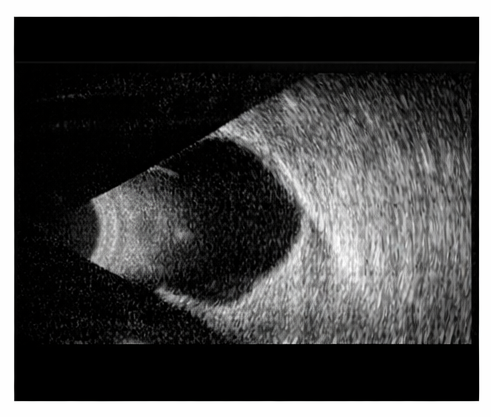
B-scan ultrasonography showed localized excavation of the posterior segment corresponding to the coloboma, without signs of retinal detachment or vitreous hemorrhage.
Systemic Evaluation
Given the potential association of coloboma with systemic syndromes, a comprehensive systemic examination was conducted. Audiological screening, cardiovascular examination, and basic laboratory investigations were within normal limits, and no systemic anomalies were identified.
Diagnosis
Based on clinical presentation and multimodal imaging findings, a definitive diagnosis of unilateral choroidal coloboma of the right eye was established.
Management and Outcome
Management Strategy
As there was no active complication such as retinal detachment or choroidal neovascularization, conservative management was advised. The patient was counseled regarding the congenital nature of the condition, the nonprogressive baseline visual impairment, and the potential risk of future complications.
Prophylactic laser photocoagulation was not performed, as there were no peripheral retinal breaks or areas of significant vitreoretinal traction. The patient was advised to avoid activities associated with ocular trauma and to report immediately if symptoms such as flashes, floaters, or sudden visual loss occurred.
Low-vision aids were discussed, although the patient was functionally independent and declined intervention at the time.
Follow-Up and Outcome
The patient was reviewed at 6-month intervals. At one-year follow-up, visual acuity remained stable, and repeat fundus examination and OCT showed no progression or development of complications. The patient continues under long-term surveillance.
Discussion
Choroidal coloboma is a developmental defect arising from abnormal embryogenesis of the eye. The inferonasal predilection reflects the site of embryonic fissure closure. Visual outcomes vary depending on macular involvement, optic nerve anomalies, and secondary complications.
One of the most serious complications associated with choroidal coloboma is retinal detachment, reported in up to 40% of affected eyes. Abnormal vitreoretinal adhesions at the coloboma margins and thin retinal tissue predispose to retinal breaks. Regular follow-up and patient education are therefore essential.
OCT has emerged as a valuable tool in assessing macular structure, detecting early complications, and guiding prognosis. While no definitive treatment exists for choroidal coloboma itself, advances in vitreoretinal surgery have improved outcomes in cases complicated by retinal detachment.
Genetic counseling may be considered in bilateral cases or when associated systemic abnormalities are present. Multidisciplinary evaluation is essential in syndromic presentations.
Conclusion
Choroidal coloboma is a rare congenital posterior segment anomaly that can lead to lifelong visual impairment and predispose patients to sight-threatening complications. Early diagnosis, multimodal imaging, patient counseling, and long-term ophthalmic surveillance are critical to preserving visual function and ensuring timely management of complications.
References
- Onwochei BC, et al. Ocular colobomata. Surv Ophthalmol. 2000;45(3):175–194.
- Gopal L, et al. Retinal detachment in eyes with choroidal coloboma. Ophthalmology. 1998;105(5):804–809.
- Khan AO. Ocular coloboma. Curr Opin Ophthalmol. 2007;18(5):370–375.
- Brodsky MC. Congenital optic disk anomalies. Surv Ophthalmol. 1994;39(2):89–112.
Read more such content on @ Hidoc Dr | Medical Learning App for Doctors
Recommended News For You
Recommended Articles For You
Featured News
Featured Articles
Featured Events
Featured KOL Videos
1.
Breast density changes over time are associated with an increased risk of cancer.
2.
How the metastasis of cancer is driven by cellular plasticity.
3.
Belzutifan Combo Could be Second-Line Option in Advanced RCC
4.
Radioligand Shows Promise in Metastatic Hormone-Sensitive Prostate Cancer
5.
Research indicates that extended PSMA therapy is safe and effective for patients with prostate cancer.
1.
Essential Oncology Updates: Diagnosis, Treatment Advances, Tools & Education Resources
2.
Unlocking the Secrets of Hemoglobin: How It Works to Keep Us Healthy
3.
Uncovering the Mystery of the Sessile Serrated Adenoma
4.
Biomimetic Nanovesicles Target Senescent-Escape Cancer Stem Cells in Breast Cancer
5.
The Science Behind Thrombolytic Drugs and Their Benefits
1.
International Lung Cancer Congress®
2.
Genito-Urinary Oncology Summit 2026
3.
Future NRG Oncology Meeting
4.
ISMB 2026 (Intelligent Systems for Molecular Biology)
5.
Annual International Congress on the Future of Breast Cancer East
1.
From Relapse to Remission: Mapping the Treatment Journey in Adult R/R-Cell ALL - Part 2
2.
An Eagles View - Evidence-based Discussion on Iron Deficiency Anemia- Panel Discussion
3.
Molecular Contrast: EGFR Axon 19 vs. Exon 21 Mutations - Part VI
4.
The Reign of the CROWN Trial and the Dawn of a New Era in Frontline Management
5.
Dacomitinib Case Presentation: Baseline Treatment and Current Status

Address :
Hidoc Dr. Inc. | Delaware C Corp | 1309 Coffeen Ave. Suite 1200, Sheridan WY, 82801
Phone :
+1-415-463-3094
Email :
anishagadia@hidoc.co
© Copyright 2026 Hidoc Dr. Inc.
Terms & Conditions - LLP | Inc. | Privacy Policy - LLP | Inc. | Account Deactivation

To get started please enter your email ID
Welcome to Hidoc Dr.
Join to enhance your clinical skills and gain specialized in-depth medical knowledge