Lung Fibrosis: Clinical Presentation, Diagnostic Evaluation, Management, and Outcomes – A Case Report
OthersPage Navigation
Abstract
Lung fibrosis represents a group of chronic interstitial lung disorders characterized by progressive scarring of lung parenchyma, leading to impaired gas exchange, reduced lung compliance, and respiratory failure in advanced stages. Etiologies include idiopathic, autoimmune, occupational, drug-induced, and post-infectious causes. Patients typically present with exertional dyspnea, chronic dry cough, and declining exercise tolerance. High-resolution computed tomography (HRCT) plays a central role in diagnosis, supported by pulmonary function testing and, in selected cases, histopathological confirmation. This case report describes a middle-aged patient with progressive fibrotic interstitial lung disease, highlighting clinical features, diagnostic workup, management strategies, and functional outcomes, with emphasis on early recognition and longitudinal follow-up.
Introduction
Pulmonary fibrosis is a pathological process characterized by excessive deposition of extracellular matrix within the interstitium of the lung, resulting in irreversible architectural distortion. It encompasses a heterogeneous spectrum of disorders, including idiopathic pulmonary fibrosis (IPF), connective tissue disease–associated interstitial lung disease (CTD-ILD), hypersensitivity pneumonitis, and drug- or radiation-induced fibrosis.
The clinical importance of lung fibrosis lies in its progressive nature and poor prognosis if untreated. Many forms present insidiously, leading to delayed diagnosis and advanced disease at presentation. Recent advances in imaging, molecular understanding, and antifibrotic therapy have transformed disease management, emphasizing the importance of early detection and risk stratification. This report presents a case of progressive fibrotic lung disease in an adult patient, illustrating diagnostic challenges, therapeutic decision-making, and outcomes.
Case Report
Patient History
A 58-year-old male presented to the pulmonary medicine outpatient clinic with complaints of progressive shortness of breath on exertion for approximately one year. Initially, dyspnea was noted during brisk walking but gradually progressed to limiting routine household activities. He also reported a persistent dry cough without hemoptysis. There was no history of fever, chest pain, wheezing, or acute respiratory episodes.
The patient was a retired factory worker with a 25-year occupational exposure to metal dust and fumes. He was a former smoker with a 15 pack-year history, having quit 10 years earlier. There was no known history of connective tissue disease, long-term medication use (including amiodarone or chemotherapy), or prior pulmonary tuberculosis. Family history was negative for interstitial lung disease.
Clinical Examination
On general examination, the patient appeared comfortable at rest but became visibly dyspneic on minimal exertion.

Oxygen saturation was 94% on room air at rest, dropping to 88% after a six-minute walk. Digital clubbing was present bilaterally. There was no cyanosis, peripheral edema, or lymphadenopathy.
Chest examination revealed bilateral fine end-inspiratory “Velcro-like” crackles predominantly at the lung bases. Cardiovascular examination was unremarkable, with no signs of right heart failure. Other systemic examinations were within normal limits.
Clinical Evaluation
Differential Diagnosis
Based on history and examination, the following differential diagnoses were considered:
• Idiopathic pulmonary fibrosis
• Chronic hypersensitivity pneumonitis
• Occupational lung disease–related fibrosis
• Connective tissue disease–associated interstitial lung disease
• Post-infectious pulmonary fibrosis
The absence of systemic autoimmune symptoms and the presence of occupational exposure guided further evaluation toward fibrotic interstitial lung disease.
Investigations
Pulmonary Function Tests (PFTs)
Spirometry demonstrated a restrictive ventilatory defect with reduced forced vital capacity (FVC) at 62% of predicted. Total lung capacity was reduced, and diffusing capacity for carbon monoxide (DLCO) was significantly decreased at 48% of predicted, indicating impaired gas exchange.
High-Resolution Computed Tomography (HRCT) Chest
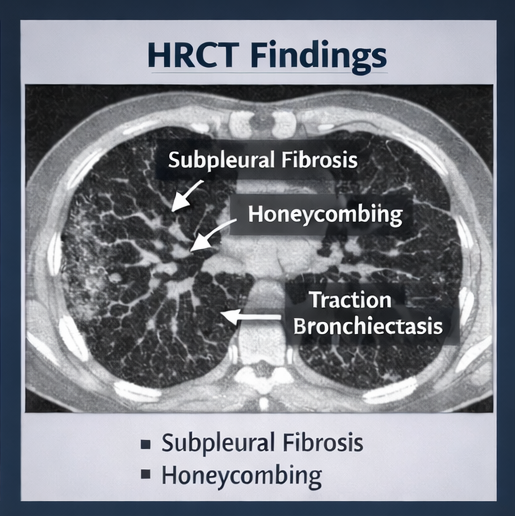
HRCT of the chest revealed bilateral, subpleural, and basal-predominant reticular opacities with traction bronchiectasis. Areas of honeycombing were noted in the posterior basal segments. There was minimal ground-glass opacity and no significant nodularity or mosaic attenuation. These findings were consistent with a usual interstitial pneumonia (UIP) pattern.
Laboratory Investigations
Baseline hematological and biochemical parameters were within normal limits. Autoimmune serology, including antinuclear antibody (ANA), rheumatoid factor, anti-CCP, and myositis panel, was negative. Inflammatory markers were not elevated.
Six-Minute Walk Test (6MWT)
The patient walked 380 meters, with a significant drop in oxygen saturation during exertion, supporting functional impairment.
Based on clinical, radiological, and functional findings, a diagnosis of fibrotic interstitial lung disease with a UIP pattern, most consistent with idiopathic pulmonary fibrosis, was made. Lung biopsy was not pursued due to classical imaging features and patient preference.
Management and Outcome
Treatment Planning
Given the progressive nature of fibrotic lung disease and radiological UIP pattern, antifibrotic therapy was initiated. The patient was counseled extensively regarding disease prognosis, treatment goals, potential adverse effects, and the importance of adherence and follow-up.
Medical Management
The patient was started on antifibrotic therapy with pirfenidone, initiated at a low dose and gradually titrated to the recommended maintenance dose.
Liver function tests were monitored regularly. Symptomatic management included:
• Short-acting bronchodilators for exertional dyspnea
• Pulmonary rehabilitation to improve exercise tolerance

• Vaccination against influenza and pneumococcus
Supplemental oxygen therapy was not required at rest but was advised during exertion if desaturation worsened.
Follow-Up and Outcome
At three-month follow-up, the patient reported stabilization of breathlessness with improved confidence in daily activities. Liver function tests remained within normal limits. Pulmonary rehabilitation improved exercise endurance.
At six-month follow-up, FVC showed minimal decline (<5%), and DLCO remained stable. HRCT did not demonstrate significant radiological progression. The patient continued antifibrotic therapy with good tolerance.
At one-year follow-up, the disease remained clinically stable with preserved quality of life. The patient was counseled regarding long-term monitoring and the potential future need for oxygen therapy or advanced interventions if disease progression occurred.
Discussion
Pulmonary fibrosis is a chronic, progressive condition with variable clinical course depending on etiology and pattern of lung involvement. UIP-pattern fibrosis, particularly in idiopathic pulmonary fibrosis, is associated with poorer prognosis compared to other interstitial lung diseases.
Early recognition of symptoms such as exertional dyspnea and dry cough is critical. Physical findings like digital clubbing and basal crackles, though subtle, are valuable clinical clues. HRCT is the cornerstone of diagnosis, often obviating the need for invasive lung biopsy when classic patterns are present.
Antifibrotic agents such as pirfenidone and nintedanib have been shown to slow the rate of lung function decline but do not reverse established fibrosis. Therefore, treatment goals focus on disease stabilization, symptom control, and preservation of quality of life. Pulmonary rehabilitation and supportive care play an essential adjunctive role.
Regular follow-up with serial pulmonary function testing and clinical assessment is necessary to detect progression early and adjust management. Lung transplantation may be considered in advanced cases for eligible patients.
Conclusion
Lung fibrosis is a serious interstitial lung disorder with significant morbidity and mortality if left untreated. This case highlights the importance of early diagnosis through clinical suspicion and HRCT imaging, timely initiation of antifibrotic therapy, and comprehensive patient education. Although fibrosis is irreversible, appropriate management can stabilize disease progression and improve functional outcomes. Long-term monitoring remains essential due to the unpredictable disease course.
References
- Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011. https://pubmed.ncbi.nlm.nih.gov/21471066/
- Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir Med. 2018. https://pubmed.ncbi.nlm.nih.gov/29154106/
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011. https://pubmed.ncbi.nlm.nih.gov/21481713/
- Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014. https://pubmed.ncbi.nlm.nih.gov/24836310/
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2011. https://pubmed.ncbi.nlm.nih.gov/21506734/
- Meyer KC. Diagnosis and management of interstitial lung disease. Transl Respir Med. 2014. https://pubmed.ncbi.nlm.nih.gov/25852826/
Read more such content on @ Hidoc Dr | Medical Learning App for Doctors
Recommended News For You
Recommended Articles For You
Featured News
Featured Articles
Featured Events
Featured KOL Videos
1.
Does pollution cause cancer?
2.
AI is equally capable of reading breast cancer scans as human radiologists.
3.
EVP Beats Cisplatin for Resectable MIBC
4.
New research points out a promising strategy for treating metastatic medulloblastoma
5.
Academics + Pharma = Big Bucks; New CAR-T Warnings; Patients Seek Cancer Tests.
1.
A Closer Look at Breast Cancer: Examining the Ultrasound Images
2.
Unlocking the Secrets of Oral Cancer Staging: A New Approach to Early Detection
3.
Impact of Hormone Therapy Cessation on Tumor Growth: Case Study of Ki-67 Reduction
4.
Unraveling the Mysteries of Lymphoma: A Journey into the Unknown
5.
Refining AML Survival: Prognostic Factors, Therapies, and Stem Cell Strategies Reviewed
1.
International Lung Cancer Congress®
2.
Genito-Urinary Oncology Summit 2026
3.
Future NRG Oncology Meeting
4.
ISMB 2026 (Intelligent Systems for Molecular Biology)
5.
Annual International Congress on the Future of Breast Cancer East
1.
Navigating the Complexities of Ph Negative ALL - Part III
2.
A Comprehensive Guide to First Line Management of ALK Positive Lung Cancer - Part VIII
3.
Management of 1st line ALK+ mNSCLC (CROWN TRIAL Update)
4.
Expert Group meeting with the management of EGFR mutation positive NSCLC - Part III
5.
Virtual Case Study on Pedal Edema and Triple Vessel Disease - An Initiative by Hidoc Dr.

Address :
Hidoc Dr. Inc. | Delaware C Corp | 1309 Coffeen Ave. Suite 1200, Sheridan WY, 82801
Phone :
+1-415-463-3094
Email :
anishagadia@hidoc.co
© Copyright 2026 Hidoc Dr. Inc.
Terms & Conditions - LLP | Inc. | Privacy Policy - LLP | Inc. | Account Deactivation

To get started please enter your email ID
Welcome to Hidoc Dr.
Join to enhance your clinical skills and gain specialized in-depth medical knowledge