Osteogenesis Imperfecta: Clinical Presentation, Diagnostic Evaluation, Management, and Outcomes – A Case Report
OthersPage Navigation
Abstract
Osteogenesis imperfecta (OI) is a rare inherited connective tissue disorder characterized by bone fragility, recurrent fractures, and variable skeletal and extraskeletal manifestations. It is primarily caused by mutations affecting type I collagen synthesis or structure. Clinical severity ranges from mild forms with few fractures to severe, life-threatening variants. Common features include blue sclerae, dentinogenesis imperfecta, hearing loss, short stature, and skeletal deformities. Diagnosis is largely clinical, supported by radiographic findings and genetic testing when available. Management is multidisciplinary and focuses on fracture prevention, orthopedic care, pharmacologic therapy such as bisphosphonates, rehabilitation, and patient education. This case report describes a young patient with osteogenesis imperfecta, highlighting clinical features, diagnostic workup, management strategy, and short-term outcomes. Early diagnosis and comprehensive care are essential to improve mobility, reduce fracture risk, and enhance quality of life.
Introduction
Osteogenesis imperfecta is a heterogeneous group of genetic disorders characterized by increased bone fragility due to defects in type I collagen. The majority of cases are caused by autosomal dominant mutations in the COL1A1 or COL1A2 genes, although autosomal recessive forms have also been described. The estimated incidence is approximately 1 in 15,000–20,000 live births.
Clinically, OI presents with recurrent fractures following minimal or no trauma, skeletal deformities, growth retardation, and characteristic extraskeletal features such as blue sclerae, dentinogenesis imperfecta, and hearing impairment. The disease is classified into several types based on clinical severity, radiographic findings, and genetic etiology. Despite advances in supportive care and pharmacologic therapy, OI remains a lifelong condition requiring long-term follow-up and multidisciplinary management.
Case Report
Patient History
A 9-year-old male child was brought to the pediatric outpatient clinic with a history of multiple fractures since early childhood. The parents reported that the child had sustained at least five fractures involving the long bones of the upper and lower limbs following trivial trauma, such as minor falls. The first fracture occurred at the age of 18 months.
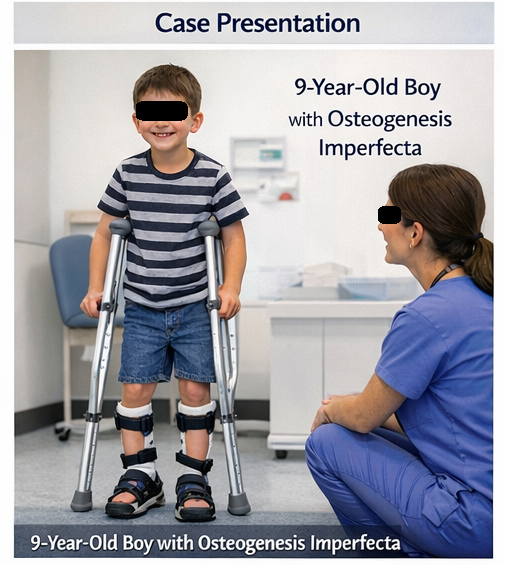
There was no history of non-accidental injury. The child was born at term with no perinatal complications. A family history revealed that the patient’s father had sustained multiple fractures during childhood, suggesting a possible inherited disorder. There was no history of chronic illness, prolonged medication use, or nutritional deficiencies.
Clinical Examination
On physical examination, the child had short stature for age and mild bowing of the lower limbs. Blue discoloration of the sclerae was noted bilaterally. Dental examination revealed opalescent teeth suggestive of dentinogenesis imperfecta.
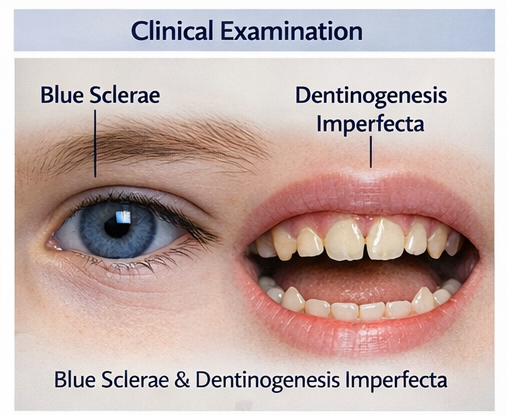
Joint hypermobility was present, but neurological examination was normal.
There were no signs of acute fracture at presentation. Hearing assessment by bedside screening did not reveal obvious deficits. Based on the clinical history and examination findings, a provisional diagnosis of osteogenesis imperfecta was considered.
Clinical Evaluation
Differential Diagnosis
The differential diagnosis for recurrent fractures in childhood includes:
- Osteogenesis imperfecta
- Nutritional rickets
- Child abuse (non-accidental injury)
- Juvenile osteoporosis
- Metabolic bone disorders
A structured evaluation was undertaken to establish the diagnosis.
Investigations
- Radiographic evaluation: X-rays of the long bones showed generalized osteopenia, cortical thinning, and evidence of healed fractures with mild deformity.
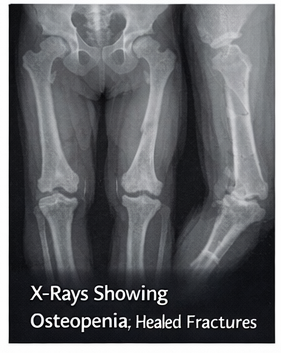
- Biochemical tests: Serum calcium, phosphate, alkaline phosphatase, and vitamin D levels were within normal limits, ruling out metabolic bone disease.
- Genetic testing: Molecular analysis revealed a pathogenic mutation in the COL1A1 gene, confirming the diagnosis of osteogenesis imperfecta type I.
Management and Outcome
Management Strategy
The patient was managed using a multidisciplinary approach involving pediatrics, orthopedics, physiotherapy, and dentistry.
Management included:
- Pharmacologic therapy: Intravenous bisphosphonate therapy was initiated to improve bone density and reduce fracture risk.
- Orthopedic care: Bracing was advised to support weight-bearing and prevent deformities. Surgical intervention was not required at this stage.
- Physiotherapy: A supervised rehabilitation program focusing on muscle strengthening, balance, and safe mobility was started.
- Supportive care: Calcium and vitamin D supplementation were provided to ensure optimal bone health.
- Patient and family education: Counseling regarding fracture prevention, safe physical activity, and genetic nature of the disease was provided.
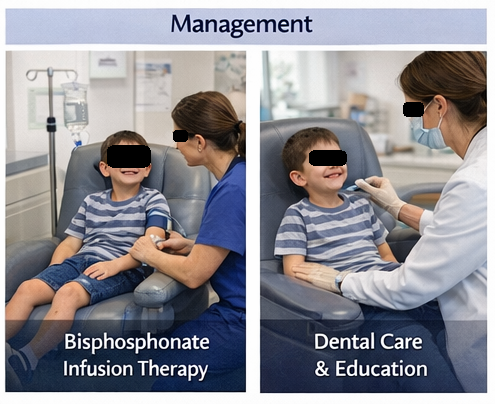
Follow-Up and Outcome
At six-month follow-up, the patient had not sustained any new fractures and demonstrated improved mobility and confidence during physical activity. Repeat radiographs showed improvement in bone mineralization. The child continued to attend regular physiotherapy sessions and remained compliant with treatment.
The family was advised regarding long-term follow-up, hearing evaluation at regular intervals, and dental care for dentinogenesis imperfecta. Genetic counseling was also offered to the parents.
Discussion
Osteogenesis imperfecta represents a broad spectrum of inherited connective tissue disorders with highly variable clinical severity, ranging from mild forms with few fractures to severe, life-threatening presentations with profound skeletal deformities. Type I OI, the mildest and most frequently encountered form, is typically characterized by blue sclerae, normal or near-normal stature, dentinogenesis imperfecta, ligamentous laxity, and recurrent fractures that may occur even with minimal trauma. Early and accurate diagnosis is critical not only to identify affected individuals but also to distinguish OI from other causes of childhood fractures, including metabolic bone disorders, nutritional deficiencies, and non-accidental injury. Misdiagnosis can lead to inappropriate management and increased risk of complications.
Pharmacologic therapy, particularly with bisphosphonates, has revolutionized the management of OI by improving bone mineral density, reducing the frequency and severity of fractures, and enhancing overall skeletal strength. While bisphosphonates do not cure the underlying collagen defect, they provide significant clinical benefit when combined with a comprehensive, multidisciplinary care plan. Long-term management emphasizes optimizing functional independence, preventing deformities, minimizing pain, and supporting physical, social, and psychological development. This approach involves close coordination among pediatricians, orthopedic surgeons, physiotherapists, geneticists, and other healthcare professionals. With sustained, individualized care, patients with OI can experience improved mobility, reduced fracture risk, and a markedly enhanced quality of life, even in the presence of a lifelong genetic condition.
Conclusion
Osteogenesis imperfecta is a heterogeneous genetic disorder primarily characterized by increased bone fragility, recurrent fractures, and varying degrees of skeletal deformity, often accompanied by multisystem involvement such as blue sclerae, dentinogenesis imperfecta, hearing loss, and ligamentous laxity. Early recognition is crucial and relies on careful clinical assessment, detailed family history, and supportive radiologic findings, with molecular genetic testing increasingly playing a key role in confirming the diagnosis and defining disease subtype. Prompt diagnosis allows timely initiation of management strategies aimed at fracture prevention, optimization of bone health, and monitoring of extra-skeletal complications. Effective care requires a multidisciplinary approach that integrates pharmacologic therapy such as bisphosphonates, orthopedic interventions for fracture management and deformity correction, structured rehabilitation and physiotherapy programs, and comprehensive patient and family education. Long-term follow-up focusing on mobility, pain control, growth, and psychosocial support is essential. With appropriate, individualized, and sustained care, many patients with mild to moderate forms of osteogenesis imperfecta can achieve good functional independence, reduced fracture burden, and a significantly improved quality of life.
References
- The Lancet – Osteogenesis imperfecta: Pathogenesis and clinical management.
- Journal of Bone and Mineral Research – Bisphosphonate therapy in osteogenesis imperfecta
- American Journal of Medical Genetics – Genetic basis of osteogenesis imperfecta
- Pediatric Clinics of North America – Evaluation of recurrent fractures in children
- Orphanet Journal of Rare Diseases – Multidisciplinary management of osteogenesis imperfecta
Read more such content on @ Hidoc Dr | Medical Learning App for Doctors
Recommended News For You
Recommended Articles For You
Featured News
Featured Articles
Featured Events
Featured KOL Videos
1.
Remote monitoring can improve recovery from cancer surgery
2.
Intractable cancers may respond better to treatment when using new radiation and high-performance computing.
3.
A Win for AI in Cancer; 2025's Transformative Drugs; FDA Clarifies 'Underway' Trials
4.
Conditional EU Nod for Weekly Pill in Pediatric Glioma
5.
high response rate when using a bispecific antibody to treat R/R multiple myeloma.
1.
The Technological Revolution in Precision Oncology and Tumor Microenvironment Therapy
2.
The Role of the Oncology Pharmacist: From Treatment to Trials and Beyond
3.
Unlocking the Secrets of Neutrophils: Exploring Their Role in Immune Defense
4.
New Hope for Rectal Cancer Patients: Breakthrough Drug Shows Promising Results
5.
Unveiling the Mystery of Echinocyte: A Closer Look at the Unique Red Blood Cell
1.
International Lung Cancer Congress®
2.
Genito-Urinary Oncology Summit 2026
3.
Future NRG Oncology Meeting
4.
ISMB 2026 (Intelligent Systems for Molecular Biology)
5.
Annual International Congress on the Future of Breast Cancer East
1.
Role of Nimotuzumab in Management of Nasopharyngeal Cancer
2.
The Landscape of First-Line Treatment for Urothelial Carcinoma- The Conclusion
3.
Pazopanib Takes Center Stage in Managing Renal Cell Carcinoma - Part III
4.
A Comprehensive Guide to First Line Management of ALK Positive Lung Cancer - Part IV
5.
An Eagles View - Evidence-based Discussion on Iron Deficiency Anemia- Panel Discussion

Address :
Hidoc Dr. Inc. | Delaware C Corp | 1309 Coffeen Ave. Suite 1200, Sheridan WY, 82801
Phone :
+1-415-463-3094
Email :
anishagadia@hidoc.co
© Copyright 2026 Hidoc Dr. Inc.
Terms & Conditions - LLP | Inc. | Privacy Policy - LLP | Inc. | Account Deactivation

To get started please enter your email ID
Welcome to Hidoc Dr.
Join to enhance your clinical skills and gain specialized in-depth medical knowledge