Acromegaly: Clinical Presentation, Diagnostic Evaluation, Management, and Outcomes – A Case Report
OthersPage Navigation
Abstract
Acromegaly is a rare, chronic endocrine disorder characterized by excessive secretion of growth hormone (GH), most commonly due to a pituitary adenoma. The condition leads to progressive somatic disfigurement, metabolic dysfunction, and multisystem complications. Because of its insidious onset and slow progression, diagnosis is often delayed, increasing the risk of morbidity and mortality.
We report the case of a 45-year-old male presenting with progressive enlargement of facial features, acral overgrowth, and metabolic disturbances. Clinical suspicion, supported by biochemical evaluation and imaging, confirmed the diagnosis of GH-secreting pituitary adenoma. The patient underwent transsphenoidal surgical resection followed by medical therapy, resulting in significant biochemical and symptomatic improvement.
This case highlights the importance of early recognition of subtle clinical features, timely diagnostic workup, and a multidisciplinary approach in the management of acromegaly to optimize long-term outcomes.
Introduction
Acromegaly is a hormonal disorder resulting from chronic hypersecretion of growth hormone, typically caused by a benign pituitary adenoma. Elevated GH levels stimulate hepatic production of insulin-like growth factor-1 (IGF-1), which mediates most of the clinical manifestations.
The condition predominantly affects middle-aged adults and is associated with increased morbidity due to cardiovascular, metabolic, and respiratory complications.
The pathophysiology involves:
- Excess GH secretion independent of normal hypothalamic regulation
- Elevated IGF-1 levels leading to tissue overgrowth
- Progressive organomegaly and metabolic dysregulation
Key etiological factors include:
- Pituitary somatotroph adenoma (most common)
- Rare ectopic GH or GHRH-secreting tumors
Risk factors are not clearly defined; however, genetic syndromes such as multiple endocrine neoplasia type 1 (MEN1) may predispose individuals.
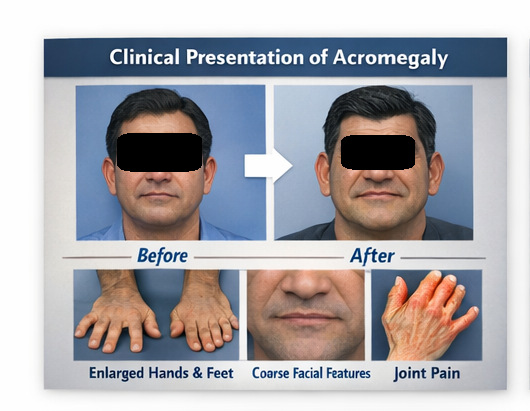
Clinically, acromegaly presents with:
- Enlargement of hands and feet
- Coarsening of facial features
- Soft tissue swelling
- Joint pain and arthropathy
- Metabolic abnormalities such as diabetes mellitus
Due to its gradual progression, diagnosis is often delayed by several years.
Case Report
Patient History
A 45-year-old male presented to the endocrinology clinic with complaints of:
- Progressive enlargement of hands and feet over 5 years
- Increasing shoe and ring size
- Facial changes including broadening of the nose and jaw
- Excessive sweating and fatigue
- Intermittent headaches
The patient also reported:
- Snoring and disturbed sleep
- Joint pain, particularly in the knees and wrists
There was no history of visual disturbances, seizures, or prior endocrine disorders.
Past medical history revealed recently diagnosed hypertension and impaired glucose tolerance. There was no significant family history of endocrine diseases.
Clinical Examination
On physical examination:
- The patient had coarse facial features with frontal bossing
- Enlarged nose, lips, and prognathism were evident
- Macroglossia was present
- Hands and feet were visibly enlarged with thickened skin
Vital signs:
- Blood pressure: 150/95 mmHg
- Body mass index (BMI): 29 kg/m²
Systemic findings included:
- Soft tissue swelling
- Mild cardiomegaly on clinical assessment
Neurological examination was unremarkable, with no visual field defects detected on confrontation testing.
Clinical Evaluation
Differential Diagnosis
The following conditions were considered:
- Acromegaly
- Hypothyroidism (myxedema features)
- Paget’s disease of bone
- Gigantism (less likely due to adult onset)
- Pseudoacromegaly (insulin resistance syndromes)
The progressive acral enlargement and facial changes strongly suggested acromegaly.
Investigations
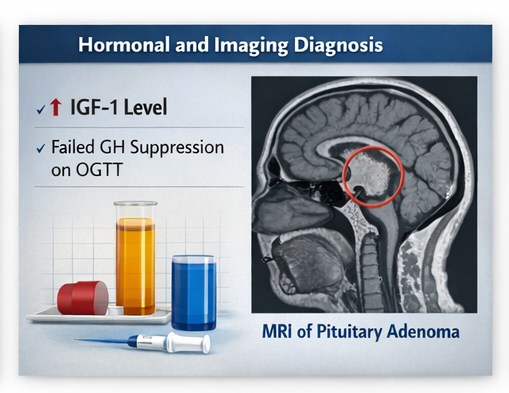
Biochemical Assessment
- Serum IGF-1 levels: markedly elevated
- Random GH levels: elevated
- Oral glucose tolerance test (OGTT): failure of GH suppression
Additional findings:
- Fasting blood glucose: elevated
- HbA1c: consistent with prediabetes
- Lipid profile: mildly deranged
Imaging
Magnetic resonance imaging (MRI) of the brain revealed:
- A well-defined pituitary macroadenoma measuring 1.5 cm
- Mild suprasellar extension
- No compression of the optic chiasm
These findings confirmed the presence of a GH-secreting pituitary tumor.
Diagnosis
Based on clinical features, biochemical evidence of GH excess, and imaging findings, a diagnosis of acromegaly secondary to pituitary macroadenoma was established.
Management and Outcome
Management Strategy
A multidisciplinary approach involving endocrinologists, neurosurgeons, and radiologists was adopted.
Surgical Treatment
The patient underwent:
- Transsphenoidal surgical resection of the pituitary adenoma
This approach is considered the first-line treatment for most patients with acromegaly.
Postoperative Care
- Monitoring of GH and IGF-1 levels
- Assessment of pituitary function
- Hormonal replacement therapy if required
Medical Therapy
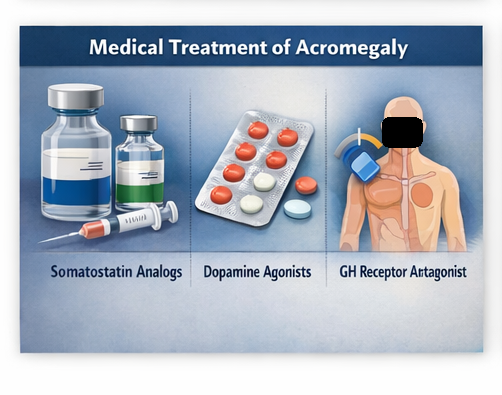
Due to residual elevated IGF-1 levels post-surgery, medical therapy was initiated:
- Somatostatin analogs (e.g., octreotide)
- Dopamine agonists as adjunct therapy
Follow-Up and Outcome

At 1 month:
- Significant reduction in GH levels
- Improvement in headaches and sweating
At 3 months:
- IGF-1 levels showed marked decline
- Blood pressure better controlled
At 6 months:
- Stabilization of metabolic parameters
- Reduction in soft tissue swelling
At 12 months:
- Biochemical remission achieved
- Improved quality of life
- No tumor recurrence on follow-up MRI
Discussion
Pathophysiology
Acromegaly results from prolonged exposure to elevated GH and IGF-1 levels, leading to:
- Soft tissue overgrowth
- Skeletal changes
- Metabolic dysregulation
Key mechanisms include:
- Increased protein synthesis
- Enhanced cell proliferation
- Insulin resistance
Cardiovascular complications are a major cause of morbidity, including:
- Hypertension
- Cardiomyopathy
- Arrhythmias
Diagnostic Challenges
Diagnosis is often delayed due to:
- Gradual onset of symptoms
- Overlap with common conditions such as obesity and arthritis
Key diagnostic tools include:
- Serum IGF-1 measurement (most reliable screening test)
- GH suppression test using OGTT
- MRI for tumor localization
Early diagnosis is critical to prevent irreversible complications.
Treatment Considerations
Surgical Management
- First-line therapy for most patients
- Offers rapid reduction in GH levels
- Success depends on tumor size and surgeon expertise
Medical Therapy
Indicated when:
- Surgery is contraindicated
- Residual disease persists
Options include:
- Somatostatin analogs
- GH receptor antagonists (e.g., pegvisomant)
- Dopamine agonists
Radiotherapy
- Reserved for refractory cases
- Delayed onset of action
- Risk of hypopituitarism
Complications

Potential complications of acromegaly include:
- Cardiovascular disease (leading cause of mortality)
- Type 2 diabetes mellitus
- Obstructive sleep apnea
- Osteoarthritis
- Colonic polyps
Treatment-related complications may include:
- Hypopituitarism
- Surgical risks
- Drug-related adverse effects
Prognosis
The prognosis of acromegaly has improved significantly with early diagnosis and modern treatment modalities.
Factors influencing outcomes include:
- Degree of GH control
- Tumor size and invasiveness
- Presence of comorbidities
Patients achieving biochemical remission have near-normal life expectancy.
Conclusion
Acromegaly is a chronic endocrine disorder with significant systemic implications if left untreated. This case highlights the importance of recognizing early clinical features such as acral enlargement and facial changes, which can prompt timely diagnostic evaluation.
Biochemical confirmation and imaging are essential for accurate diagnosis, while transsphenoidal surgery remains the cornerstone of treatment. Adjunct medical therapy plays a crucial role in achieving complete disease control.
A multidisciplinary approach, regular follow-up, and patient education are vital to ensure optimal outcomes and reduce long-term complications.
Early intervention not only improves clinical symptoms but also significantly enhances quality of life and survival in patients with acromegaly.
References
- Melmed S. Acromegaly pathogenesis and treatment. Journal of Clinical Investigation. https://pubmed.ncbi.nlm.nih.gov/16123994/
- Katznelson L, Laws ER Jr, Melmed S, et al. Acromegaly: Endocrine Society Clinical Practice Guideline. Journal of Clinical Endocrinology & Metabolism. https://pubmed.ncbi.nlm.nih.gov/22723309/
- Colao A, Grasso LF, Giustina A, Melmed S, Chanson P, Pereira AM, Pivonello R. Acromegaly. Lancet Diabetes Endocrinology. https://pubmed.ncbi.nlm.nih.gov/26972928/
- Giustina A, Chanson P, Bronstein MD, et al. A consensus on the diagnosis and treatment of acromegaly. Nature Reviews Endocrinology. https://pubmed.ncbi.nlm.nih.gov/28680166/
- Katznelson L. Approach to the patient with acromegaly. Journal of Clinical Endocrinology & Metabolism. https://pubmed.ncbi.nlm.nih.gov/17519315/
- Melmed S, Bronstein MD, Chanson P, et al. A consensus statement on acromegaly therapeutic outcomes. Endocrine Reviews. https://pubmed.ncbi.nlm.nih.gov/29522177/
- Ben-Shlomo A, Melmed S. Pituitary tumors in acromegaly. Endocrinology and Metabolism Clinics of North America. https://pubmed.ncbi.nlm.nih.gov/25910714/
- Holdaway IM, Rajasoorya C. Epidemiology of acromegaly. Pituitary. https://pubmed.ncbi.nlm.nih.gov/11043806/
Read more such content on @ Hidoc Dr | Medical Learning App for Doctors
Recommended News For You
Recommended Articles For You
Featured News
Featured Articles
Featured Events
Featured KOL Videos
1.
Electronic Sepsis Alerts; Reducing Plaques in Coronary Arteries
2.
Ivonescimab Tops Pembrolizumab in PD-L1-Positive, Advanced NSCLC
3.
Hereditary cancer has a rare and underreported cause.
4.
New imaging guidelines for head and neck cancers, a step toward practice change
5.
BMTs that are "half-matched" are effective in treating severe sickle cell disease.
1.
Oncolytic Adenoviruses Targeting PD-L1: Advancing Cancer Immunotherapy and Tumor Control
2.
Personalized Cancer Vaccines: The Next Frontier in Precision Oncology
3.
Essential Updates in Hematology in Daily Practice
4.
The Predictive Power of Theranostics in Palliative Neuroendocrine Tumor Management
5.
Importance of Early Detection in Oncology
1.
Asian Symposium on Advancement in Hematology and Oncology
2.
Asian Symposium on Advancement in Hematology and Oncology
3.
Asian Symposium on Advancement in Hematology and Oncology
4.
International Cancer Conference
5.
Asian Symposium on Advancement in Hematology and Oncology
1.
A Comprehensive Guide to First Line Management of ALK Positive Lung Cancer - Part VII
2.
Expert Group meeting with the management of EGFR mutation positive NSCLC - Part I
3.
Current Scenario of Cancer- The Incidence of Cancer in Men
4.
Untangling The Best Treatment Approaches For ALK Positive Lung Cancer - Part IV
5.
A New Era in Managing Cancer-Associated Thrombosis

Address :
Hidoc Dr. Inc. | Delaware C Corp | 1309 Coffeen Ave. Suite 1200, Sheridan WY, 82801
Phone :
+1-415-463-3094
Email :
anishagadia@hidoc.co
© Copyright 2026 Hidoc Dr. Inc.
Terms & Conditions - LLP | Inc. | Privacy Policy - LLP | Inc. | Account Deactivation

To get started please enter your email ID
Welcome to Hidoc Dr.
Join to enhance your clinical skills and gain specialized in-depth medical knowledge