Crystalline Corneal Dystrophy: Progressive Visual Impairment Due to Stromal Crystal Deposition – A Case Report
OthersPage Navigation
Abstract
Crystalline corneal dystrophy is a rare inherited corneal disorder characterized by progressive accumulation of cholesterol and phospholipid crystals within the corneal stroma. The condition is most commonly associated with Schnyder crystalline corneal dystrophy (SCCD), an autosomal dominant disorder caused by mutations in the UBIAD1 gene. Patients typically present with gradually progressive visual impairment, glare, photophobia, and decreased contrast sensitivity. The disease often manifests during early adulthood and progresses over several decades.
We present the case of a 42-year-old female who presented with progressive bilateral visual blurring and glare while driving at night. Slit-lamp examination revealed bilateral central stromal crystalline deposits with associated corneal haze. Genetic testing confirmed Schnyder crystalline corneal dystrophy. The patient underwent conservative management initially, followed by phototherapeutic keratectomy due to worsening visual symptoms. Significant improvement in visual acuity and quality of life was achieved following intervention.
This case highlights the importance of early recognition, genetic diagnosis, and timely management of crystalline corneal dystrophy to prevent visual disability.
Introduction
Corneal dystrophies are a heterogeneous group of inherited disorders characterized by progressive deposition of abnormal material within the cornea. These conditions are typically bilateral, symmetric, slowly progressive, and unrelated to environmental or inflammatory causes.
Crystalline corneal dystrophy, particularly Schnyder crystalline corneal dystrophy (SCCD), is a rare stromal dystrophy characterized by deposition of cholesterol and phospholipid crystals within the corneal stroma. Although crystalline deposits are considered a hallmark feature, they may not always be visible, especially in advanced stages.
The disease is inherited in an autosomal dominant pattern and is linked to mutations in the UBIAD1 gene. Corneal opacity gradually increases with age, leading to visual deterioration.
Common clinical manifestations include:
• Progressive visual blurring
• Glare sensitivity
• Photophobia
• Reduced contrast sensitivity
• Recurrent corneal erosions
• Corneal haze
The condition can significantly impair daily activities and quality of life if left untreated.
Case Report
Patient History
A 42-year-old female presented to the ophthalmology clinic with complaints of:
• Gradually progressive blurred vision in both eyes for five years
• Difficulty driving at night
• Increased glare from bright lights
• Mild photophobia
• Intermittent foreign body sensation
The patient denied ocular trauma, infection, or previous ocular surgery.
Family history revealed that her father and paternal grandmother had experienced progressive vision loss requiring corneal surgery during middle age.
Past medical history was significant for:
• Hypercholesterolemia
• Hypertension
She was receiving atorvastatin therapy for lipid control.
Clinical Examination
General Examination
Vital signs were stable.
• Blood pressure: 126/82 mmHg
• Pulse rate: 78/min
• Temperature: Afebrile
Systemic examination was otherwise unremarkable.
Ophthalmic Examination
Visual Acuity
Right eye (OD): 20/60
Left eye (OS): 20/80
Best-corrected visual acuity improved minimally with refraction.
Slit-Lamp Examination
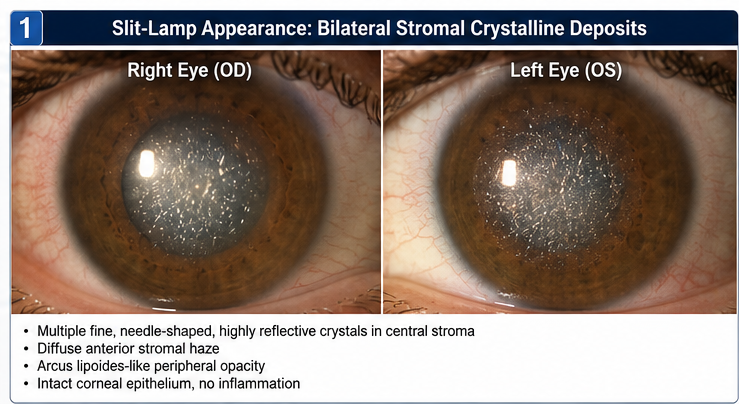
Findings included:
• Bilateral central corneal crystalline deposits
• Diffuse anterior stromal haze
• Arcus lipoides-like peripheral opacity
• Intact corneal epithelium
• No signs of inflammation
The crystalline deposits appeared as multiple fine, needle-shaped, highly reflective opacities within the central corneal stroma.
Intraocular Pressure
• Right eye: 15 mmHg
• Left eye: 16 mmHg
Fundus Examination
Posterior segment examination was normal in both eyes.
Clinical Evaluation
Differential Diagnosis
The following conditions were considered:
• Schnyder crystalline corneal dystrophy
• Bietti crystalline dystrophy
• Infectious crystalline keratopathy
• Monoclonal gammopathy-associated keratopathy
• Cystinosis
• Drug-induced crystalline deposits
Given the family history and bilateral symmetric involvement, inherited corneal dystrophy was strongly suspected.
Investigations
Corneal Imaging
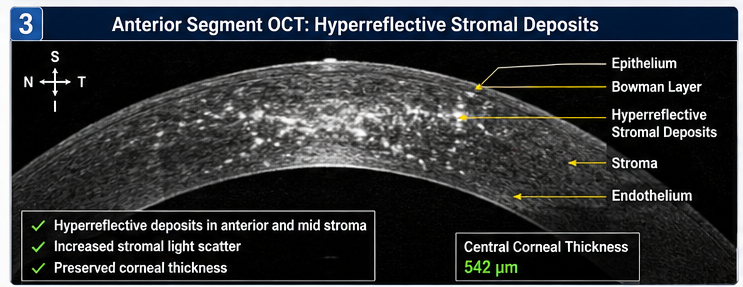
Anterior Segment Optical Coherence Tomography (AS-OCT)
AS-OCT demonstrated:
• Hyperreflective stromal deposits
• Increased stromal light scatter
• Preservation of corneal thickness
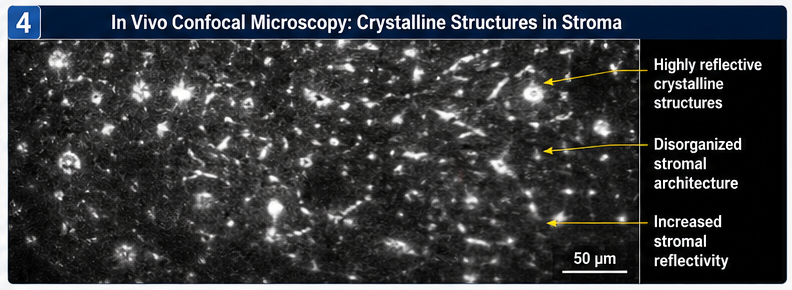
In Vivo Confocal Microscopy
Confocal imaging revealed:
• Numerous reflective crystalline structures within anterior and mid stroma
• Disorganized stromal architecture
• Increased stromal reflectivity
Laboratory Evaluation
Results showed:
• Total cholesterol: 278 mg/dL
• LDL cholesterol: 182 mg/dL
• HDL cholesterol: 42 mg/dL
• Triglycerides: 198 mg/dL
Renal and liver function tests were normal.
Serum protein electrophoresis showed no evidence of monoclonal gammopathy.
Genetic Analysis
Molecular testing identified a pathogenic mutation in the UBIAD1 gene, confirming the diagnosis of Schnyder crystalline corneal dystrophy.
Diagnosis
Based on clinical findings, imaging studies, family history, and genetic confirmation, a diagnosis of: Schnyder Crystalline Corneal Dystrophy (SCCD) was established.
Management and Outcome
Initial Management
The patient was initially managed conservatively with:
• Lubricating eye drops
• UV-protective eyewear
• Night-driving precautions
• Lipid-lowering therapy optimization
Regular follow-up visits were scheduled every six months.
Disease Progression
Over the following two years, the patient experienced:
• Increasing corneal haze
• Progressive visual decline
• Worsening glare sensitivity
• Reduced quality of life
Visual acuity deteriorated to:
• Right eye: 20/100
• Left eye: 20/120
Surgical Intervention
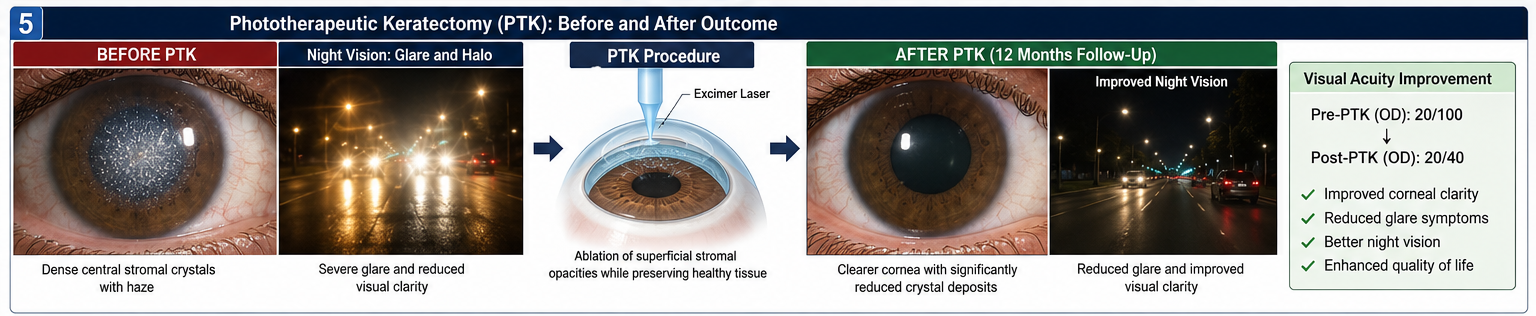
Due to worsening visual symptoms, phototherapeutic keratectomy (PTK) was performed in the right eye.
The procedure involved:
• Excimer laser removal of superficial stromal opacities
• Preservation of healthy corneal tissue
• Postoperative topical antibiotic and corticosteroid therapy
Follow-Up
At 1 Month
• Improved corneal clarity
• Reduced glare symptoms
• Visual acuity improved to 20/50
At 3 Months
• Stable corneal surface
• Significant reduction in stromal haze
• Improved night vision
At 12 Months
• Visual acuity maintained at 20/40
• No significant recurrence of symptoms
• High patient satisfaction
The fellow eye continued under observation for possible future intervention.
Discussion
Pathophysiology
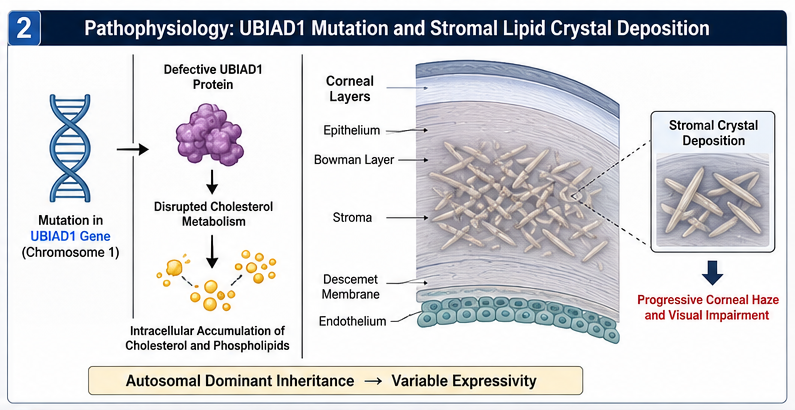
Schnyder crystalline corneal dystrophy is caused by mutations in the UBIAD1 gene located on chromosome 1.
The UBIAD1 protein is involved in cholesterol metabolism and intracellular lipid regulation. Genetic abnormalities result in abnormal deposition of cholesterol and phospholipids within corneal stromal layers.
The disease demonstrates variable expressivity, and not all patients develop visible crystalline deposits.
Clinical Features
Clinical manifestations often vary with age.
Early-stage disease may present with:
• Central crystalline deposits
• Mild visual symptoms
Advanced disease may demonstrate:
• Dense stromal haze
• Arcus lipoides
• Mid-peripheral corneal opacification
• Marked visual impairment
Patients commonly report:
• Blurred vision
• Glare
• Photophobia
• Reduced contrast sensitivity
Differential Diagnosis
Bietti Crystalline Dystrophy
Unlike SCCD, Bietti dystrophy primarily affects the retina and retinal pigment epithelium.
Infectious Crystalline Keratopathy
Usually associated with corneal infection and prior ocular surgery.
Monoclonal Gammopathy
May produce crystalline corneal deposits secondary to systemic paraproteinemia.
Cystinosis
Typically presents during childhood and is associated with systemic manifestations.
Diagnostic Considerations
Accurate diagnosis requires:
- Detailed family history
- Slit-lamp biomicroscopy
- Corneal imaging
- Lipid profile assessment
- Genetic testing
Modern imaging techniques such as confocal microscopy and AS-OCT have significantly improved diagnostic accuracy.
Treatment Options
Management depends upon disease severity.
Conservative Therapy
Includes:
• Artificial tears
• Glare protection
• Lipid control
• Periodic monitoring
Phototherapeutic Keratectomy
PTK can improve vision by removing superficial opacities and reducing irregular corneal surfaces.
Keratoplasty
Advanced cases may require:
• Deep anterior lamellar keratoplasty (DALK)
• Penetrating keratoplasty (PK)
Recurrence may occur even after corneal transplantation because the underlying genetic defect persists.
Complications
Potential complications include:
• Progressive visual impairment
• Severe corneal haze
• Recurrent corneal erosions
• Reduced quality of life
• Need for repeated surgical intervention
Prognosis
The prognosis depends on:
• Extent of corneal involvement
• Age at diagnosis
• Rate of disease progression
• Timeliness of intervention
Most patients maintain useful vision for many years with appropriate monitoring and treatment.
Advances in molecular diagnostics and corneal surgery have substantially improved long-term outcomes.
Conclusion
Crystalline corneal dystrophy is a rare inherited stromal corneal disorder characterized by progressive lipid crystal deposition and visual deterioration. This case demonstrates the importance of comprehensive ophthalmic evaluation, advanced imaging, and genetic testing in establishing an accurate diagnosis. Early recognition and appropriate intervention, including phototherapeutic keratectomy when indicated, can significantly improve visual outcomes and quality of life. Continued surveillance remains essential because disease progression and recurrence may occur despite treatment.
References
- Weiss JS, Kruth HS, Kuivaniemi H, et al. Schnyder Corneal Dystrophy. Cornea. 2008. https://pubmed.ncbi.nlm.nih.gov/18520505/
- Witsch-Baumgartner M, Gruber M, Kraft HG, et al. UBIAD1 Mutations Cause Schnyder Crystalline Corneal Dystrophy. American Journal of Human Genetics. 2007. https://pubmed.ncbi.nlm.nih.gov/17668384/
- Orr A, Dube MP, Marcadier J, et al. Mutations in UBIAD1 Cause Schnyder Crystalline Corneal Dystrophy. American Journal of Human Genetics. 2007. https://pubmed.ncbi.nlm.nih.gov/17668383
- Aldave AJ, Han J, Frausto RF. Genetics of the Corneal Dystrophies. Current Opinion in Ophthalmology. 2011. https://pubmed.ncbi.nlm.nih.gov/21378580/
Read more such content on @ Hidoc Dr | Medical Learning App for Doctors
Recommended News For You
Recommended Articles For You
Featured News
Featured Articles
Featured Events
Featured KOL Videos
1.
Novel ADC Improves Survival in Metastatic TNBC
2.
An Examine More Into the Acceptance of CRISPR/Cas9 Gene Therapy for Sickle Cell Illness.
3.
Celebrity Cancers Stoking Fear? Cisplatin Shortage Ends; Setback for Anti-TIGIT
4.
Pancreatic cancer RNA vaccine shows durable T cell immunity
5.
Healthcare in the Mix in President Biden's Farewell Address
1.
Interpreting Iron Studies: What Your Blood Results Really Mean
2.
Unveiling New Hope: Potential Therapeutic Targets in Hematological Malignancies
3.
Feline Anemia: Diagnosis and Treatment with Focus on Rasburicase Complications
4.
Andexanet for Factor Xa Inhibitor-Associated Acute Intracerebral Hemorrhage
5.
Biologic Therapies for Cutaneous Immune-Related Adverse Events in the Era of Immune Checkpoint Inhibitors
1.
Asian Symposium on Advancement in Hematology and Oncology
2.
Asian Symposium on Advancement in Hematology and Oncology
3.
Asian Symposium on Advancement in Hematology and Oncology
4.
International Cancer Conference
5.
Asian Symposium on Advancement in Hematology and Oncology
1.
Redefining Treatment Pathways in Relapsed/Refractory Adult B-Cell ALL
2.
Breaking Down PALOMA-2: How CDK4/6 Inhibitors Redefined Treatment for HR+/HER2- Metastatic Breast Cancer
3.
Untangling The Best Treatment Approaches For ALK Positive Lung Cancer - Part I
4.
Cost Burden/ Burden of Hospitalization For R/R ALL Patients
5.
Untangling The Best Treatment Approaches For ALK Positive Lung Cancer - Part VI

Address :
Hidoc Dr. Inc. | Delaware C Corp | 1309 Coffeen Ave. Suite 1200, Sheridan WY, 82801
Phone :
+1-415-463-3094
Email :
anishagadia@hidoc.co
© Copyright 2026 Hidoc Dr. Inc.
Terms & Conditions - LLP | Inc. | Privacy Policy - LLP | Inc. | Account Deactivation

To get started please enter your email ID
Welcome to Hidoc Dr.
Join to enhance your clinical skills and gain specialized in-depth medical knowledge