Polycythemia Vera Presenting with Erythrocytosis and Pruritus: A Case Report
OthersPage Navigation
Abstract
Polycythemia vera (PV) is a chronic myeloproliferative neoplasm characterized by uncontrolled erythrocyte production, most commonly due to the JAK2 V617F mutation. Increased blood viscosity predisposes patients to arterial and venous thrombosis, making early diagnosis crucial. Common symptoms include headache, dizziness, facial plethora, fatigue, and aquagenic pruritus. We report the case of a 58-year-old man who presented with recurrent headaches, generalized itching after bathing, and facial redness for three months. Investigations revealed elevated hemoglobin and hematocrit, low serum erythropoietin, positivity for the JAK2 V617F mutation, and hypercellular bone marrow with panmyelosis. The patient was diagnosed with polycythemia vera and treated successfully with therapeutic phlebotomy, low-dose aspirin, and hydroxyurea. Clinical symptoms improved significantly, and hematological parameters normalized during follow-up. This case highlights the importance of early recognition and timely treatment in preventing thrombotic complications.
Introduction
Polycythemia vera is a Philadelphia chromosome-negative myeloproliferative neoplasm characterized by excessive production of erythrocytes with variable leukocytosis and thrombocytosis. Approximately 95% of patients harbor the JAK2 V617F mutation, resulting in constitutive activation of the JAK-STAT signaling pathway and uncontrolled hematopoiesis.
The disease typically affects individuals between 50 and 70 years of age and may initially present with nonspecific symptoms such as headache, dizziness, fatigue, blurred vision, facial plethora, or aquagenic pruritus. Untreated PV significantly increases the risk of cerebrovascular accidents, myocardial infarction, deep vein thrombosis, and pulmonary embolism. Diagnosis is based on WHO criteria incorporating elevated hemoglobin or hematocrit, characteristic bone marrow findings, JAK2 mutation positivity, and low serum erythropoietin levels. Early diagnosis and maintenance of hematocrit below 45% substantially reduce thrombotic risk.
Case Report
A 58-year-old man presented with recurrent headaches, generalized itching after hot showers, dizziness, fatigue, and facial redness for three months. He denied fever, weight loss, bleeding, chest pain, or breathlessness. There was no history of smoking, chronic lung disease, cyanotic heart disease, or high-altitude residence. His medical history included well-controlled hypertension.
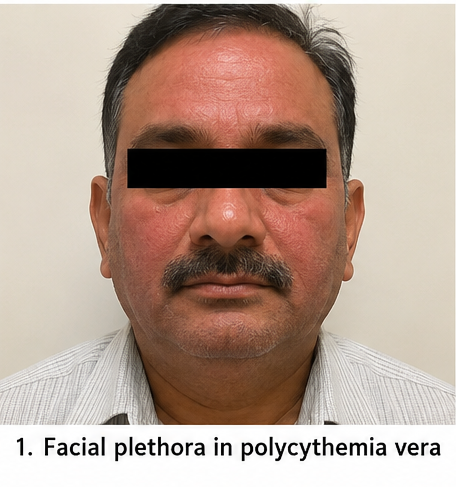
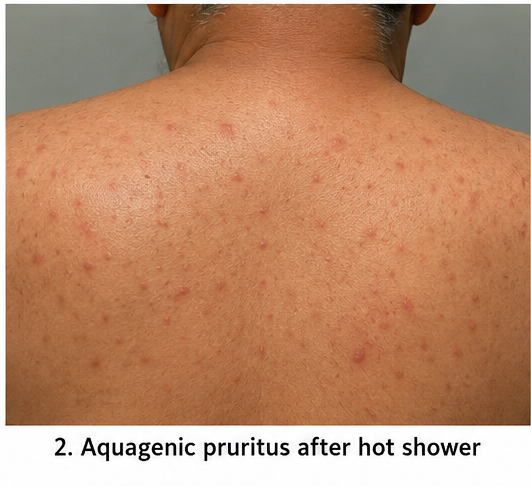
On examination, the patient was alert and hemodynamically stable. Pulse rate was 82 beats/minute, blood pressure was 148/92 mmHg, respiratory rate was 18 breaths/minute, temperature was 98.6°F, and oxygen saturation was 98% on room air. Facial plethora and mild conjunctival congestion were noted. The spleen was palpable 2 cm below the left costal margin. Cardiovascular, respiratory, abdominal, and neurological examinations were otherwise normal.
Differential diagnoses included polycythemia vera, secondary erythrocytosis due to chronic hypoxia, erythropoietin-secreting tumors, and relative polycythemia secondary to dehydration.
Laboratory investigations demonstrated:
- Hemoglobin: 19.4 g/dL
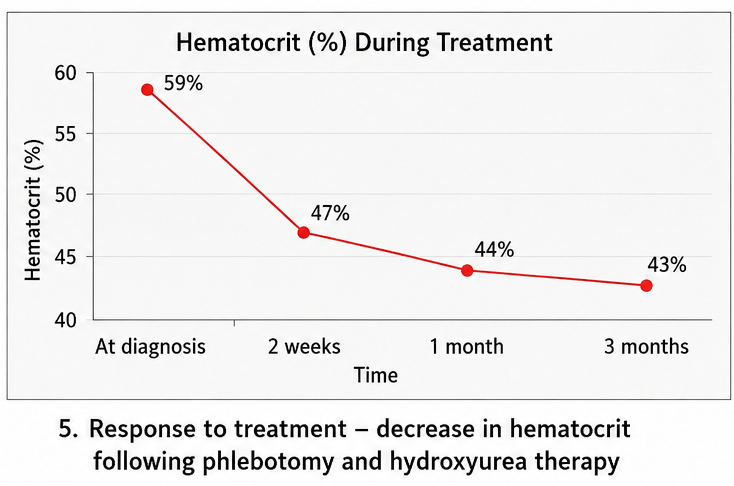
- Hematocrit: 59%
- Red blood cell count: 7.3 × 10⁶/µL
- Total leukocyte count: 13,100/mm³
- Platelet count: 548,000/mm³
- Serum erythropoietin: 2.4 mIU/mL (low)
- Liver and renal function tests: Normal
- Peripheral smear: Erythrocytosis with thrombocytosis
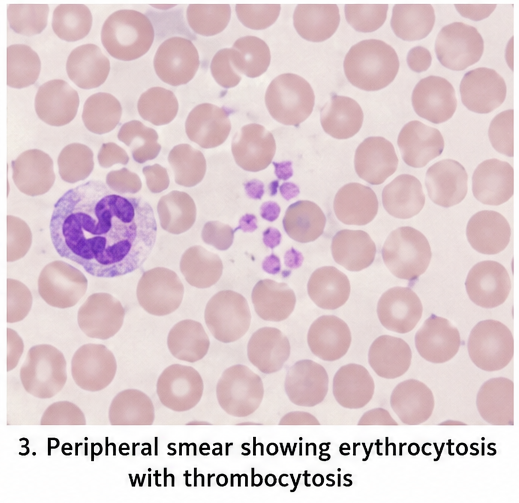
Molecular analysis detected the JAK2 V617F mutation. Ultrasonography showed mild splenomegaly.
Bone marrow biopsy revealed hypercellularity with trilineage proliferation (panmyelosis), increased erythroid precursors, and prominent mature megakaryocytes.
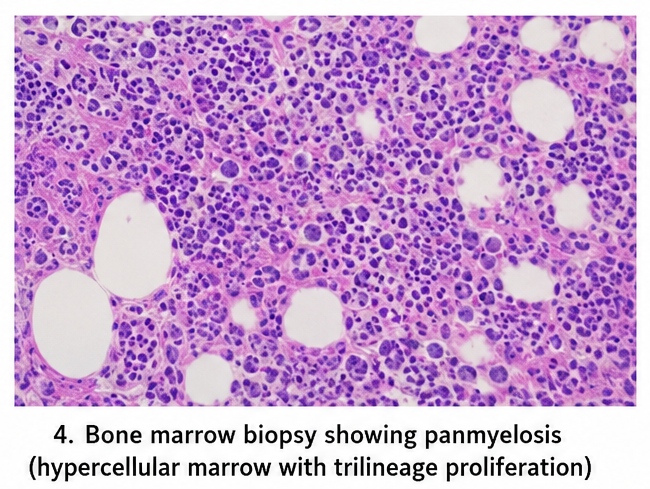
Based on WHO diagnostic criteria, a diagnosis of polycythemia vera was established.
Management and Outcome
The patient underwent weekly therapeutic phlebotomy with removal of approximately 450 mL of blood until hematocrit decreased below 45%. Medical management included low-dose aspirin (75 mg daily), hydroxyurea (500 mg twice daily), adequate hydration, and continuation of antihypertensive therapy. Lifestyle modifications and education regarding thrombotic warning signs were provided.
Within two weeks, headaches and dizziness improved considerably, while aquagenic pruritus gradually subsided over the following month. No thrombotic or hemorrhagic events occurred during treatment.
Follow-up
Two Weeks
- Reduced headache and dizziness
- Hematocrit decreased to 47%
One Month
- Aquagenic pruritus markedly improved
- Hemoglobin: 16.3 g/dL
- Hematocrit: 44%
- Platelet count: 405,000/mm³
Three Months
The patient remained asymptomatic with stable hematological parameters.
- Hemoglobin: 15.8 g/dL
- Hematocrit: 43%
- Platelet count: 372,000/mm³
Hydroxyurea and aspirin were continued with regular hematology follow-up.
Discussion
Polycythemia vera results from clonal proliferation of hematopoietic stem cells driven predominantly by JAK2 mutations. Increased red cell mass produces hyperviscosity, leading to neurological symptoms and a markedly increased risk of arterial and venous thrombosis. Aquagenic pruritus is a characteristic symptom that should raise suspicion for PV, particularly when accompanied by erythrocytosis.
The WHO diagnostic criteria facilitate accurate diagnosis by combining hematological findings, bone marrow morphology, JAK2 mutation analysis, and serum erythropoietin estimation. In this patient, all major diagnostic criteria were fulfilled, allowing prompt initiation of therapy.
Therapeutic phlebotomy remains the cornerstone of treatment and effectively reduces blood viscosity. Low-dose aspirin decreases thrombotic risk, while hydroxyurea is recommended for high-risk patients, including those older than 60 years or with previous thrombotic events. Early diagnosis and appropriate treatment significantly reduce morbidity and improve long-term survival. Lifelong monitoring is essential because some patients may progress to myelofibrosis or acute leukemia.
Prognosis
With early diagnosis and appropriate treatment, most patients with polycythemia vera achieve good long-term disease control. Regular monitoring, maintenance of hematocrit below 45%, and adherence to therapy substantially reduce thrombotic complications and improve overall quality of life.
Conclusion
Polycythemia vera should be considered in patients presenting with persistent erythrocytosis, headache, facial plethora, aquagenic pruritus, and other features suggestive of increased blood viscosity. Early recognition of these characteristic clinical manifestations, combined with appropriate laboratory investigations, is essential for establishing a timely diagnosis and preventing serious complications. Confirmation through JAK2 mutation analysis, bone marrow examination, and serum erythropoietin estimation allows accurate differentiation of polycythemia vera from secondary causes of erythrocytosis, facilitating prompt initiation of targeted therapy.
Therapeutic phlebotomy remains the cornerstone of treatment for maintaining hematocrit below the recommended target, while low-dose aspirin significantly reduces the risk of thrombotic events. In high-risk patients, cytoreductive therapy with hydroxyurea provides effective control of erythrocytosis, leukocytosis, and thrombocytosis, further minimizing disease-related complications. Regular hematological monitoring, patient education regarding symptom recognition, and long-term follow-up are equally important to ensure sustained disease control, detect progression at an early stage, and improve overall quality of life. This case highlights that early diagnosis, individualized treatment, and continuous surveillance can achieve excellent clinical outcomes while substantially reducing the risk of life-threatening vascular complications associated with polycythemia vera.
References
-
Tefferi A, Barbui T. Polycythemia Vera and Essential Thrombocythemia: 2023 Update on Diagnosis, Risk Stratification, and Management. American Journal of Hematology. 2023;98(9):1465–1481. https://pubmed.ncbi.nlm.nih.gov/37357958/
-
Arber DA, Orazi A, Hasserjian RP, et al. The 2022 WHO Classification of Hematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36:1703–1719. https://pubmed.ncbi.nlm.nih.gov/35732829/
-
Barbui T, Tefferi A, Vannucchi AM, et al. Philadelphia Chromosome-Negative Classical Myeloproliferative Neoplasms: Revised Management Recommendations from European LeukemiaNet. Leukemia. 2018;32(5):1057–1069. https://pubmed.ncbi.nlm.nih.gov/29326480/
-
Tefferi A, Barbui T. Polycythemia Vera: A Comprehensive Review and Clinical Recommendations. Mayo Clinic Proceedings. 2019;94(4):599–610. https://pubmed.ncbi.nlm.nih.gov/30947929/
-
Spivak JL. Polycythemia Vera: Myths, Mechanisms, and Management. Blood. 2019;134(4):341–352. https://pubmed.ncbi.nlm.nih.gov/31171587/
-
National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology: Myeloproliferative Neoplasms (Version 2.2025). https://www.nccn.org/professionals/physician_gls/pdf/mpn.pdf
Read more such content on @ Hidoc Dr | Medical Learning App for Doctors
Recommended News For You
Recommended Articles For You
Featured News
Featured Articles
Featured Events
Featured KOL Videos
1.
Novel ADC Improves Survival in Metastatic TNBC
2.
An Examine More Into the Acceptance of CRISPR/Cas9 Gene Therapy for Sickle Cell Illness.
3.
Celebrity Cancers Stoking Fear? Cisplatin Shortage Ends; Setback for Anti-TIGIT
4.
Pancreatic cancer RNA vaccine shows durable T cell immunity
5.
Healthcare in the Mix in President Biden's Farewell Address
1.
Interpreting Iron Studies: What Your Blood Results Really Mean
2.
Unveiling New Hope: Potential Therapeutic Targets in Hematological Malignancies
3.
Feline Anemia: Diagnosis and Treatment with Focus on Rasburicase Complications
4.
Andexanet for Factor Xa Inhibitor-Associated Acute Intracerebral Hemorrhage
5.
Biologic Therapies for Cutaneous Immune-Related Adverse Events in the Era of Immune Checkpoint Inhibitors
1.
Asian Symposium on Advancement in Hematology and Oncology
2.
Asian Symposium on Advancement in Hematology and Oncology
3.
Asian Symposium on Advancement in Hematology and Oncology
4.
International Cancer Conference
5.
Asian Symposium on Advancement in Hematology and Oncology
1.
Redefining Treatment Pathways in Relapsed/Refractory Adult B-Cell ALL
2.
Breaking Down PALOMA-2: How CDK4/6 Inhibitors Redefined Treatment for HR+/HER2- Metastatic Breast Cancer
3.
Untangling The Best Treatment Approaches For ALK Positive Lung Cancer - Part I
4.
Cost Burden/ Burden of Hospitalization For R/R ALL Patients
5.
Untangling The Best Treatment Approaches For ALK Positive Lung Cancer - Part VI

Address :
Hidoc Dr. Inc. | Delaware C Corp | 1309 Coffeen Ave. Suite 1200, Sheridan WY, 82801
Phone :
+1-415-463-3094
Email :
anishagadia@hidoc.co
© Copyright 2026 Hidoc Dr. Inc.
Terms & Conditions - LLP | Inc. | Privacy Policy - LLP | Inc. | Account Deactivation

To get started please enter your email ID
Welcome to Hidoc Dr.
Join to enhance your clinical skills and gain specialized in-depth medical knowledge